
Comparative Analysis of Transcriptional Responses to Genotoxic and Non-Genotoxic Agents in the Blood Cell Model TK6 and the Liver Model HepaRG


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
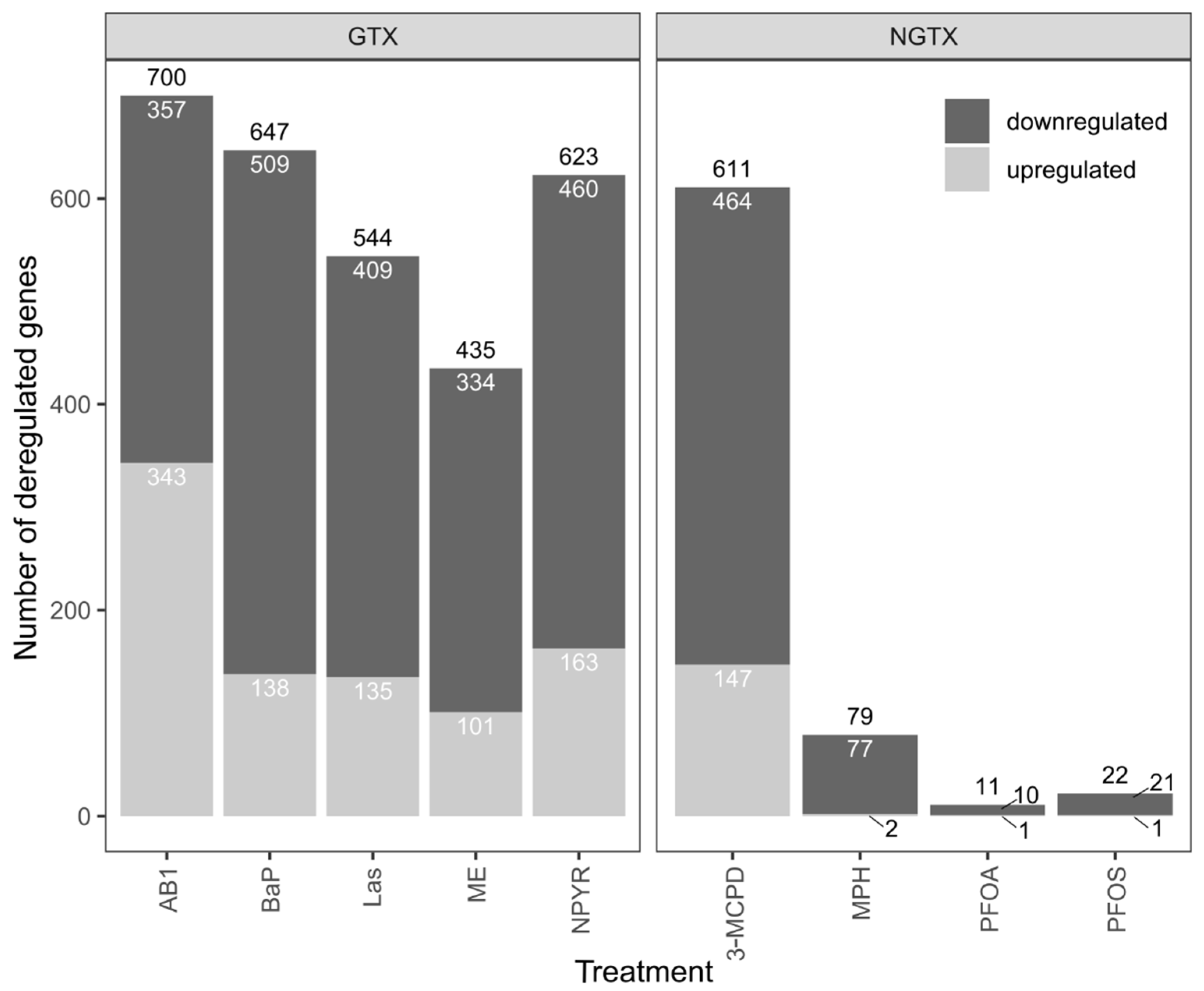
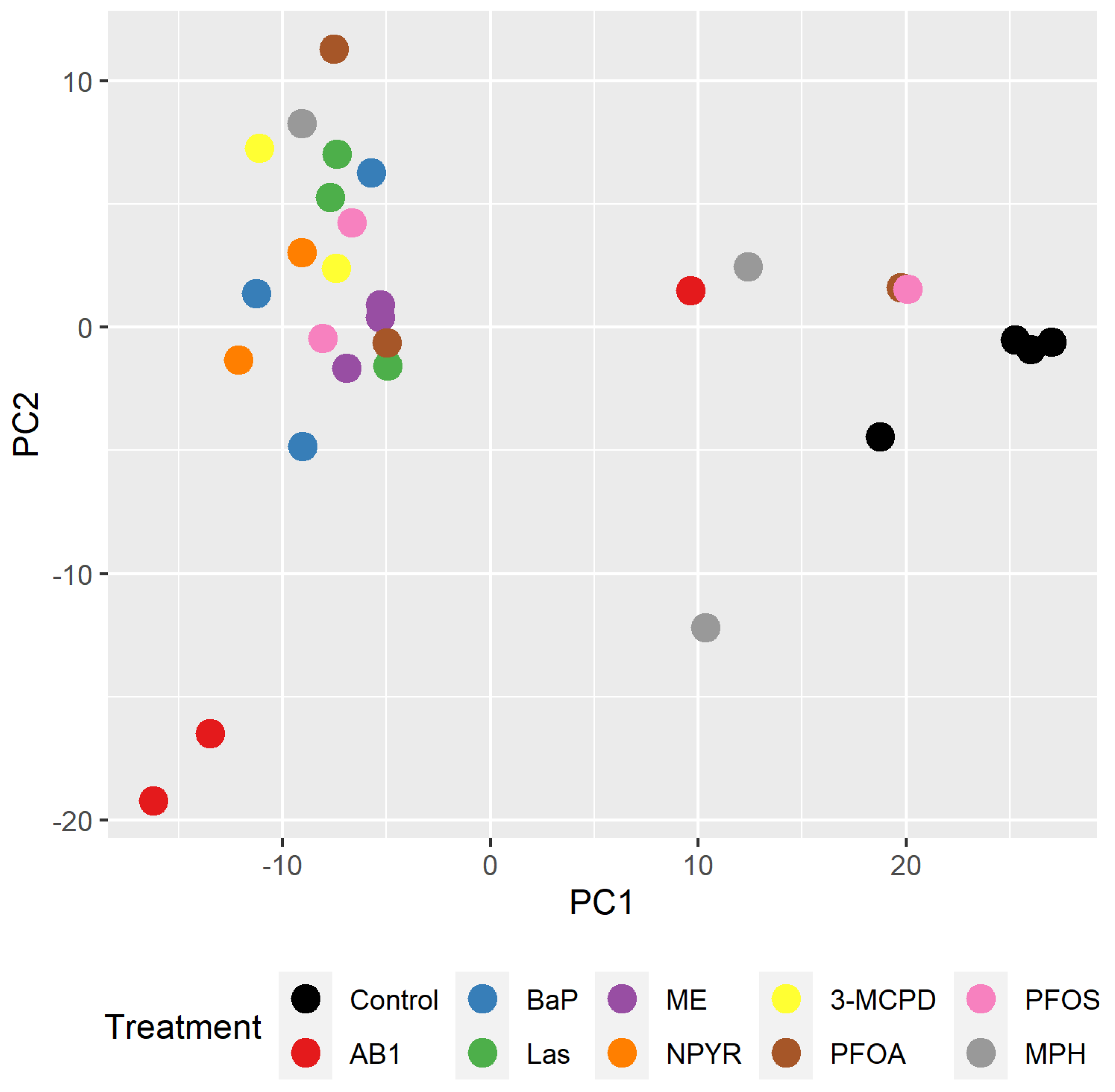
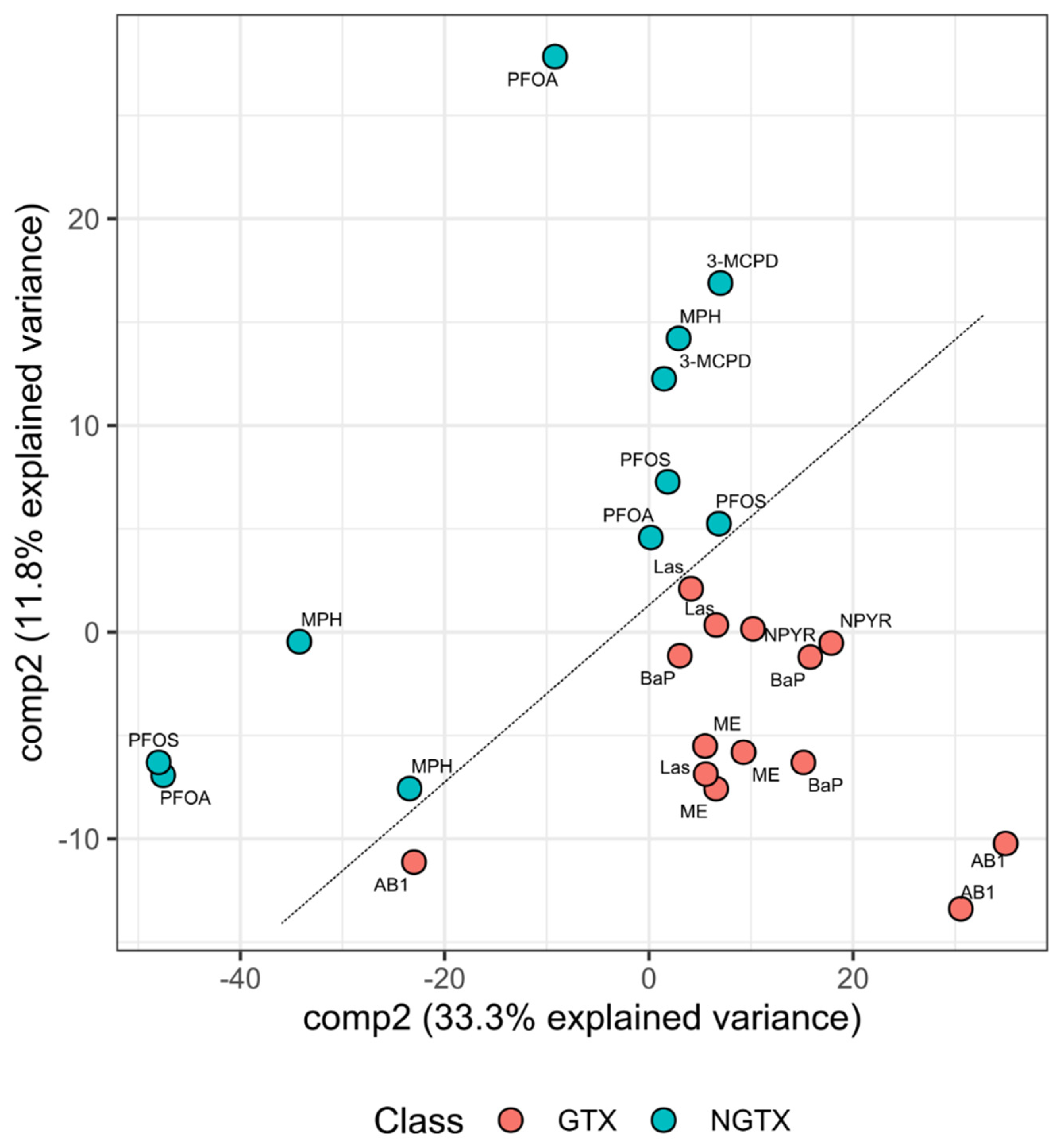
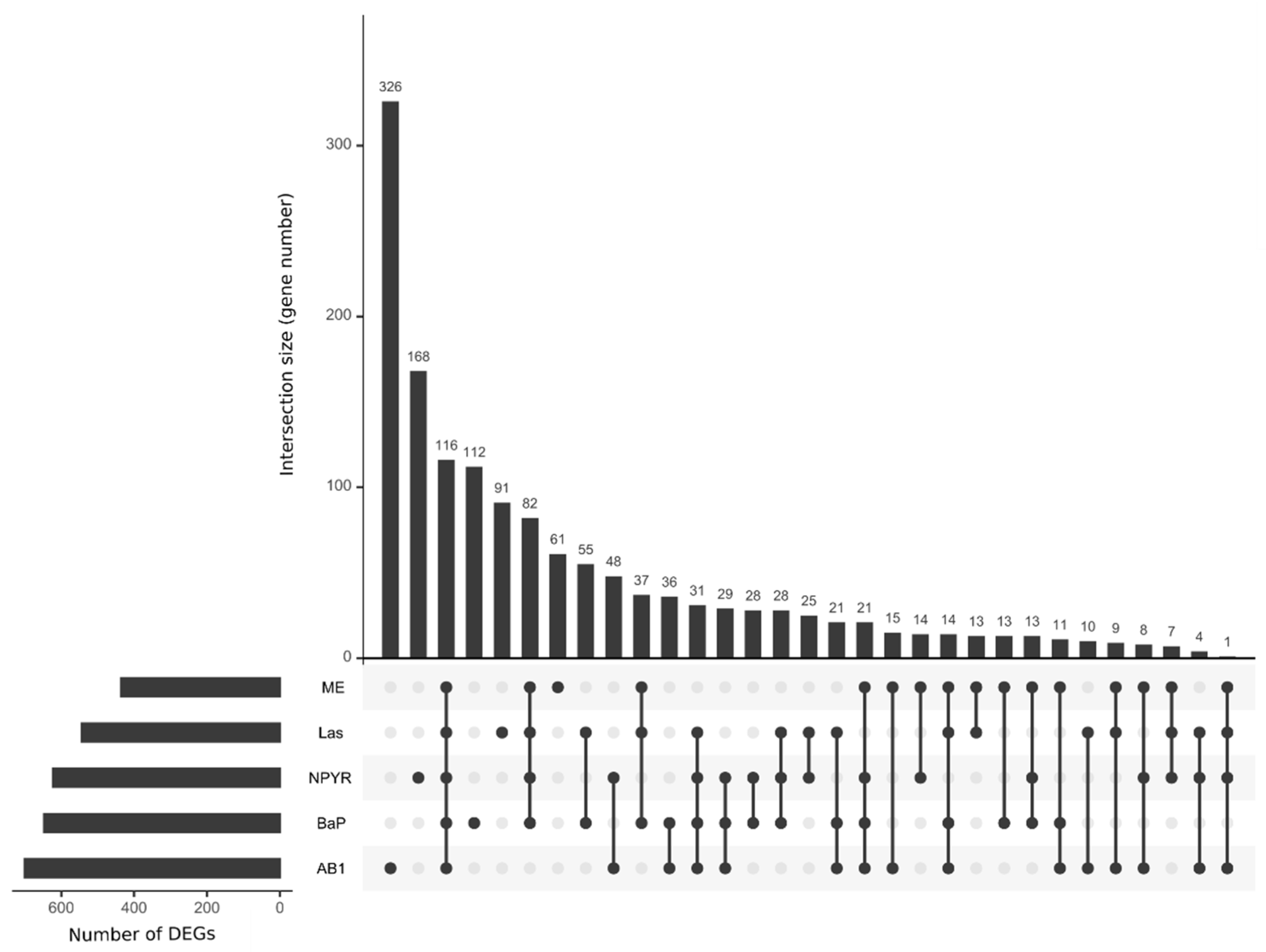
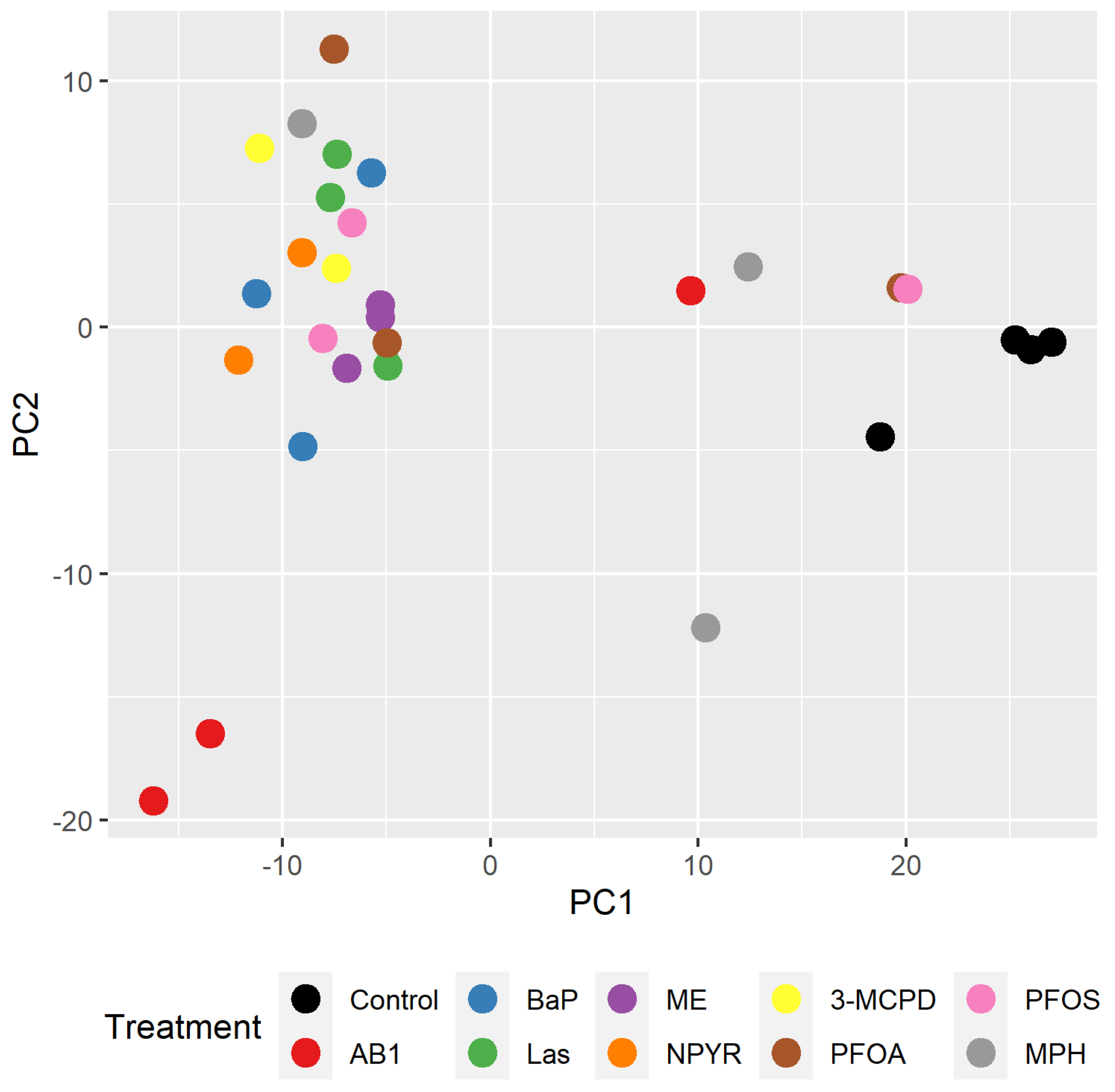
2.1. Gene Expression Analysis by RNA Sequencing in TK6 Cells
2.2. Bioinformatic Analysis of Impact of Treatments on Gene Expression in TK6 Cells
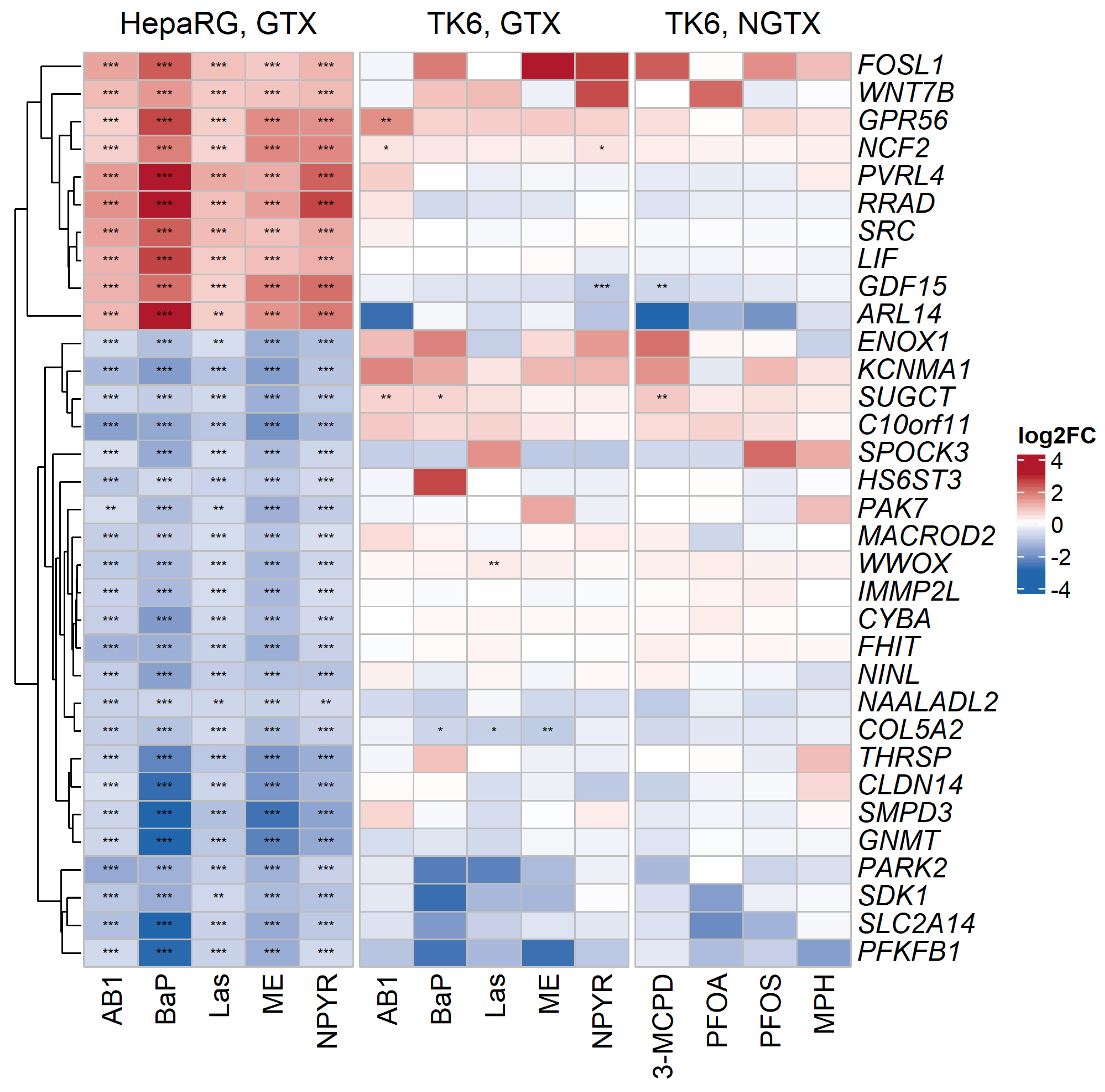
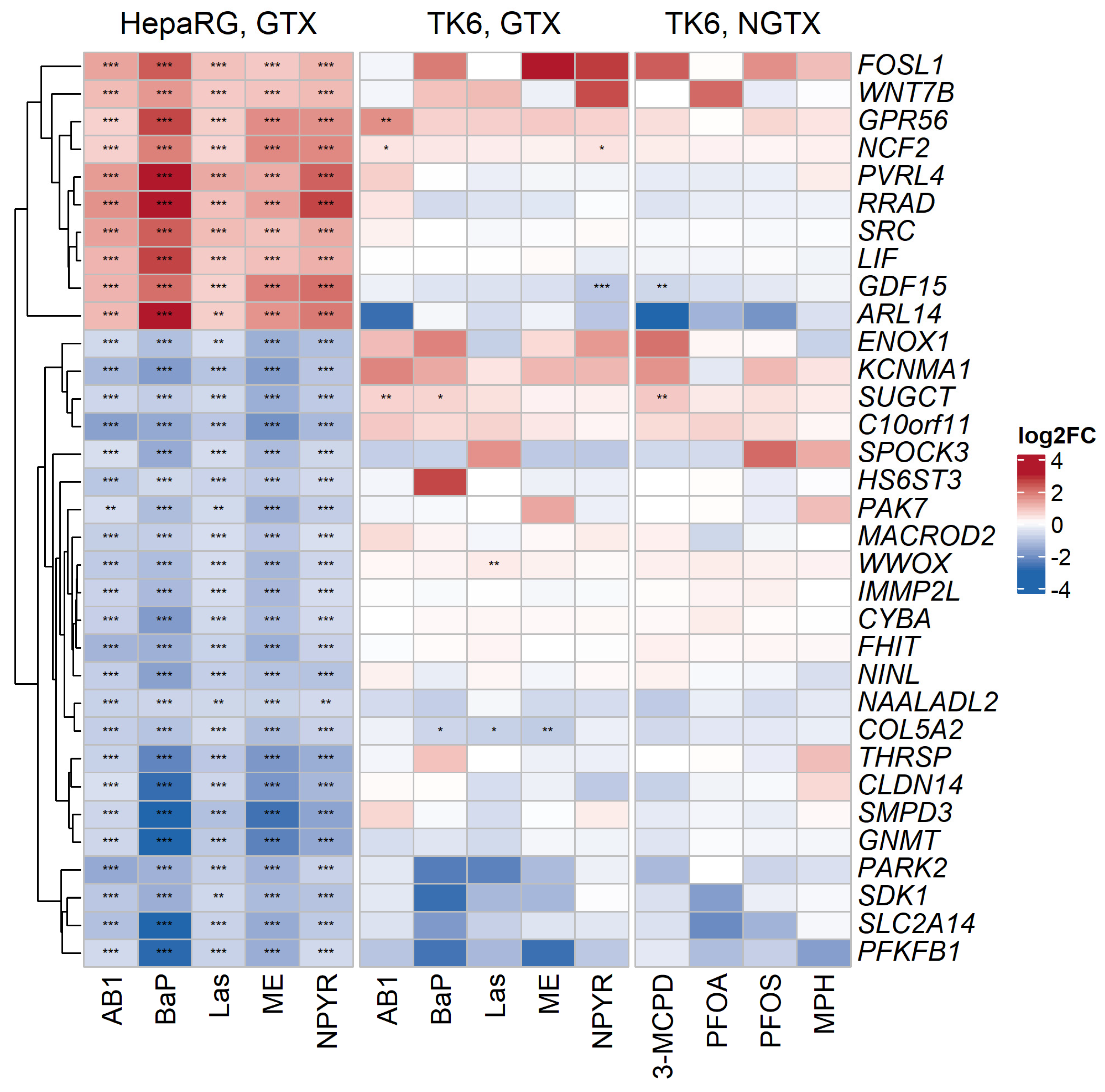
2.3. Comparison with Published Effect Marker Pattern for Genotoxicity
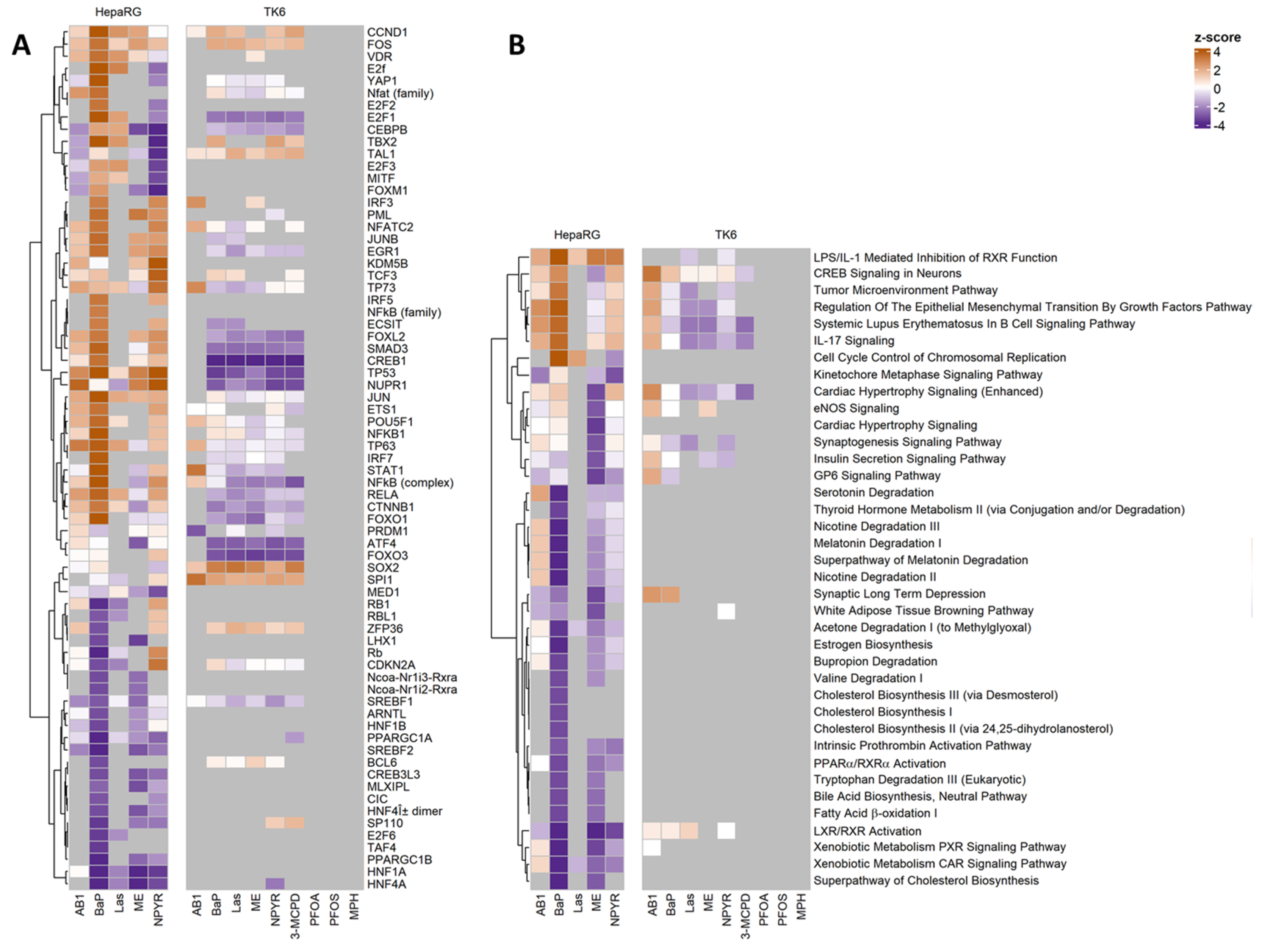
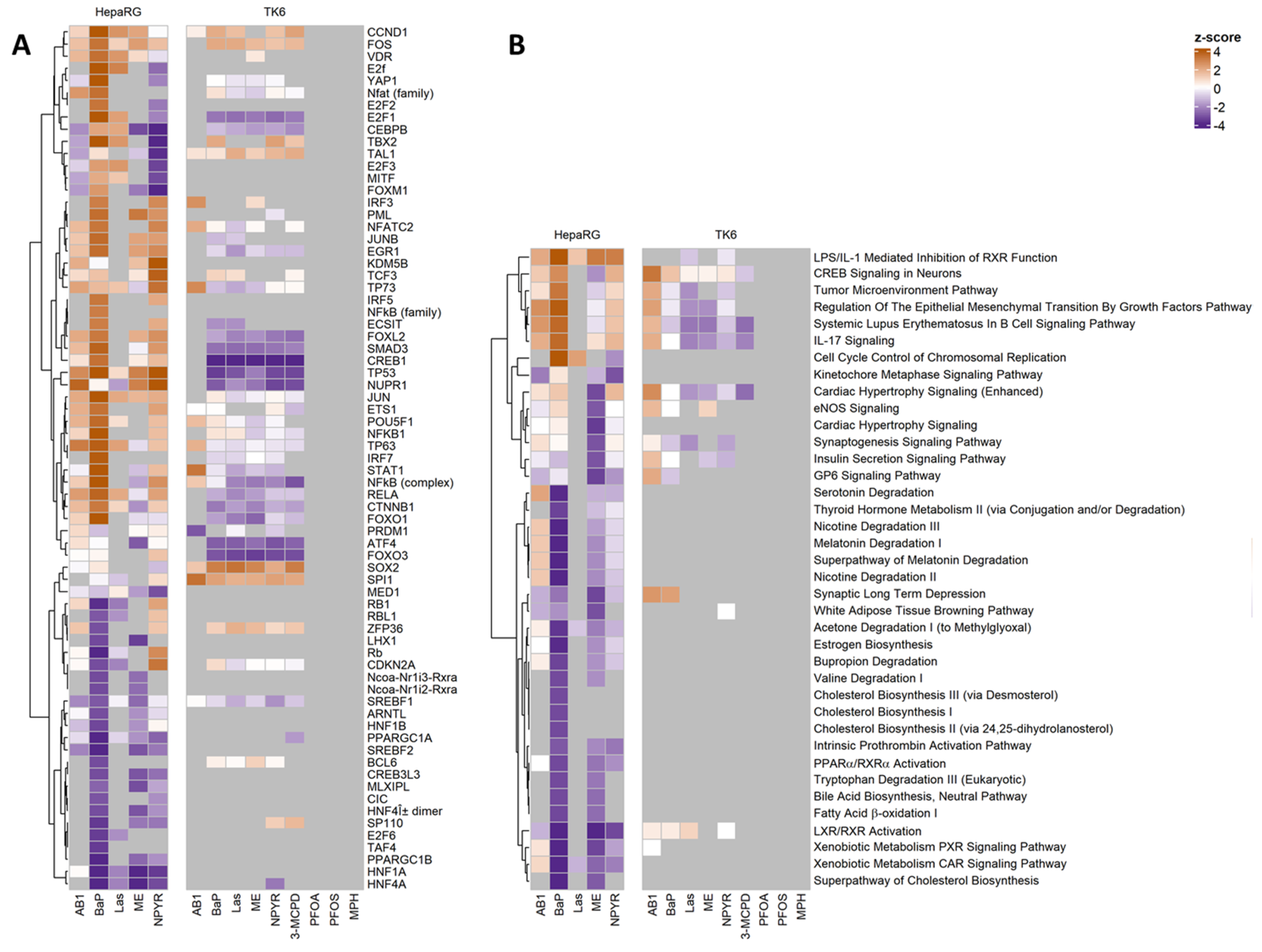
2.4. Pathways Analysis of Gene Expression Response in TK6 Cells
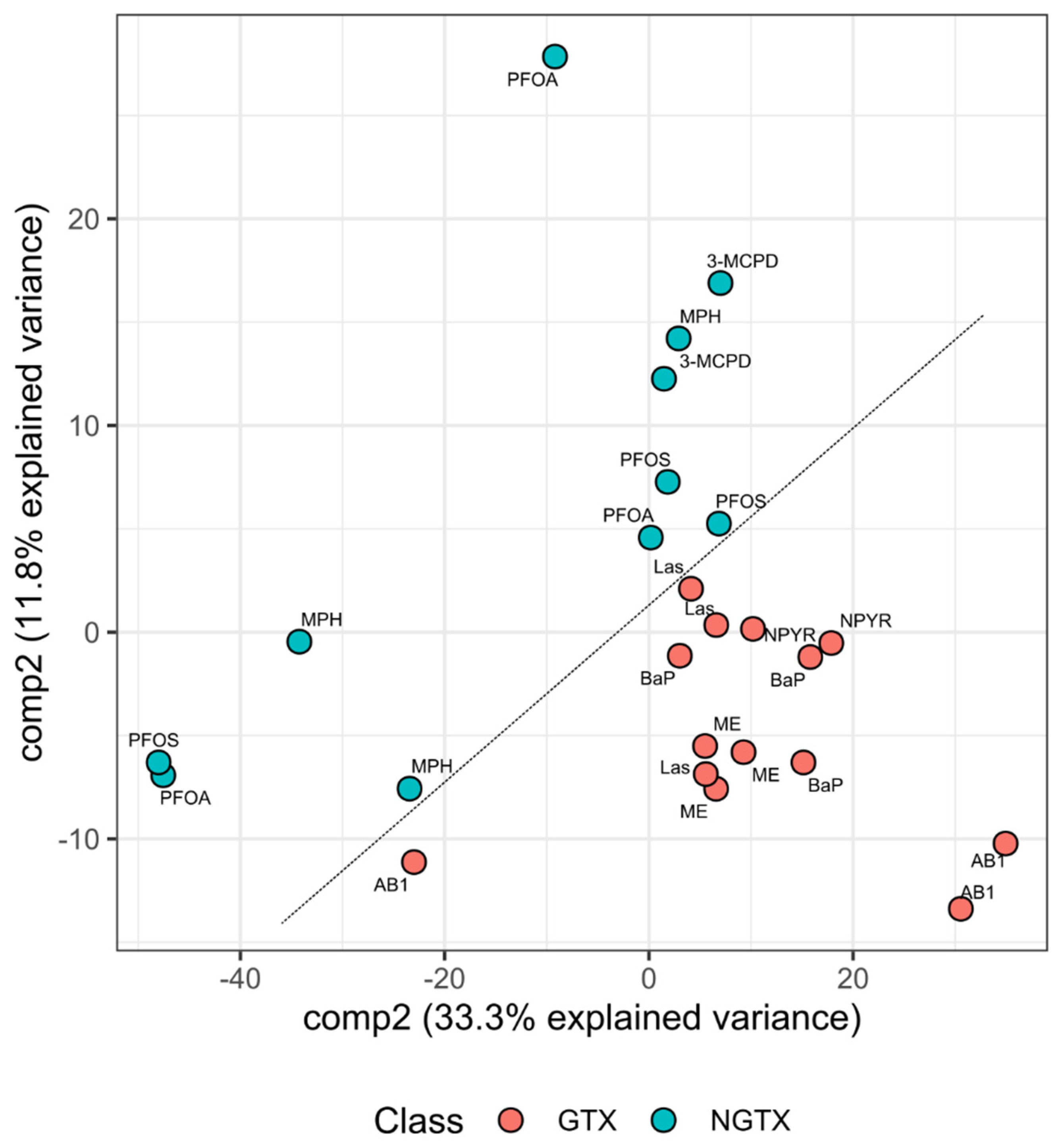
2.5. Classification of GTX and NGTX Compounds by Transcript Markers
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. RNA Isolation
4.5. RNA Sequencing
4.6. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR)
4.7. Bioinformatic Analysis and Statistics
4.8. Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corvi, R.; Madia, F. In vitro genotoxicity testing—Can the performance be enhanced? Food Chem. Toxicol. 2017, 106, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Nesslany, F. The current limitations of in vitro genotoxicity testing and their relevance to the in vivo situation. Food Chem. Toxicol. 2017, 106, 609–615. [Google Scholar] [CrossRef] [PubMed]
- David, R. The promise of toxicogenomics for genetic toxicology: Past, present and future. Mutagenesis 2020, 35, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Doktorova, T.Y.; Yildirimman, R.; Vinken, M.; Vilardell, M.; Vanhaecke, T.; Gmuender, H.; Bort, R.; Brolen, G.; Holmgren, G.; Li, R.; et al. Transcriptomic responses generated by hepatocarcinogens in a battery of liver-based in vitro models. Carcinogenesis 2013, 34, 1393–1402. [Google Scholar] [CrossRef] [Green Version]
- Kossler, N.; Matheis, K.A.; Ostenfeldt, N.; Bach Toft, D.; Dhalluin, S.; Deschl, U.; Kalkuhl, A. Identification of specific mRNA signatures as fingerprints for carcinogenesis in mice induced by genotoxic and nongenotoxic hepatocarcinogens. Toxicol. Sci. 2015, 143, 277–295. [Google Scholar] [CrossRef] [Green Version]
- Magkoufopoulou, C.; Claessen, S.M.H.; Tsamou, M.; Jennen, D.G.J.; Kleinjans, J.C.S.; van Delft, J.H.M. A transcriptomics-based in vitro assay for predicting chemical genotoxicity in vivo. Carcinogenesis 2012, 33, 1421–1429. [Google Scholar] [CrossRef]
- Rieswijk, L.; Brauers, K.J.; Coonen, M.L.; van Breda, S.G.; Jennen, D.G.; Kleinjans, J.C. Evaluating microRNA profiles reveals discriminative responses following genotoxic or non-genotoxic carcinogen exposure in primary mouse hepatocytes. Mutagenesis 2015, 30, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, A.; Arand, M.; Epe, B.; Guth, S.; Jahnke, G.; Lampen, A.; Martus, H.-J.; Monien, B.; Rietjens, I.M.C.M.; Schmitz-Spanke, S.; et al. Mode of action-based risk assessment of genotoxic carcinogens. Arch. Toxicol. 2020, 94, 1787–1877. [Google Scholar] [CrossRef]
- McMullen, P.D.; Pendse, S.N.; Black, M.B.; Mansouri, K.; Haider, S.; Andersen, M.E.; Clewell, R.A. Addressing systematic inconsistencies between in vitro and in vivo transcriptomic mode of action signatures. Toxicol. Vitr. 2019, 58, 1–12. [Google Scholar] [CrossRef]
- Canzler, S.; Schor, J.; Busch, W.; Schubert, K.; Rolle-Kampczyk, U.E.; Seitz, H.; Kamp, H.; von Bergen, M.; Buesen, R.; Hackermüller, J. Prospects and challenges of multi-omics data integration in toxicology. Arch. Toxicol. 2020, 94, 371–388. [Google Scholar] [CrossRef] [Green Version]
- Kinaret, P.A.S.; Serra, A.; Federico, A.; Kohonen, P.; Nymark, P.; Liampa, I.; Ha, M.K.; Choi, J.S.; Jagiello, K.; Sanabria, N.; et al. Transcriptomics in toxicogenomics, part I: Experimental design, technologies, publicly available data, and regulatory aspects. Nanomaterials 2020, 10, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx-Stoelting, P.; Braeuning, A.; Buhrke, T.; Lampen, A.; Niemann, L.; Oelgeschlaeger, M.; Rieke, S.; Schmidt, F.; Heise, T.; Pfeil, R.; et al. Application of omics data in regulatory toxicology: Report of an international BfR expert workshop. Arch. Toxicol. 2015, 89, 2177–2184. [Google Scholar] [CrossRef]
- Kreuzer, K.; Frenzel, F.; Lampen, A.; Braeuning, A.; Böhmert, L. Transcriptomic effect marker patterns of genotoxins—A comparative study with literature data. J. Appl. Toxicol. 2019, 40, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Kreuzer, K.; Böhmert, L.; Alhalabi, D.; Buhrke, T.; Lampen, A.; Braeuning, A. Identification of a transcriptomic signature of food-relevant genotoxins in human HepaRG hepatocarcinoma cells. Food Chem. Toxicol. 2020, 140, 111297. [Google Scholar] [CrossRef]
- Williams, A.; Buick, J.K.; Moffat, I.; Swartz, C.D.; Recio, L.; Hyduke, D.R.; Li, H.H.; Fornace, A.J., Jr.; Aubrecht, J.; Yauk, C.L. A predictive toxicogenomics signature to classify genotoxic versus non-genotoxic chemicals in human TK6 cells. Data Brief 2015, 5, 77–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryndyak, V.; Kindrat, I.; Dreval, K.; Churchwell, M.I.; Beland, F.A.; Pogribny, I.P. Effect of aflatoxin B1, benzo[a]pyrene, and methapyrilene on transcriptomic and epigenetic alterations in human liver HepaRG cells. Food Chem. Toxicol. 2018, 121, 214–223. [Google Scholar] [CrossRef]
- Ates, G.; Mertens, B.; Heymans, A.; Verschaeve, L.; Milushev, D.; Vanparys, P.; Roosens, N.H.C.; De Keersmaecker, S.C.J.; Rogiers, V.; Doktorova, T.Y. A novel genotoxin-specific qPCR array based on the metabolically competent human HepaRG™ cell line as a rapid and reliable tool for improved in vitro hazard assessment. Arch. Toxicol. 2018, 92, 1593–1608. [Google Scholar] [CrossRef]
- Georgiadis, P.; Hebels, D.G.; Valavanis, I.; Liampa, I.; Bergdahl, I.A.; Johansson, A.; Palli, D.; Chadeau-Hyam, M.; Chatziioannou, A.; Jennen, D.G.; et al. Omics for prediction of environmental health effects: Blood leukocyte-based cross-omic profiling reliably predicts diseases associated with tobacco smoking. Sci. Rep. 2016, 6, 20544. [Google Scholar] [CrossRef] [Green Version]
- Skopek, T.R.; Liber, H.L.; Penman, B.W.; Thilly, W.G. Isolation of a human lymphoblastoid line heterozygous at the thymidine kinase locus: Possibility for a rapid human cell mutation assay. Biochem. Biophys. Res. Commun. 1978, 84, 411–416. [Google Scholar] [CrossRef]
- Li, C.Q.; Robles, A.I.; Hanigan, C.L.; Hofseth, L.J.; Trudel, L.J.; Harris, C.C.; Wogan, G.N. Apoptotic signaling pathways induced by nitric oxide in human lymphoblastoid cells expressing wild-type or mutant p53. Cancer Res. 2004, 64, 3022–3029. [Google Scholar] [CrossRef] [Green Version]
- Oka, H.; Ouchida, M.; Kondo, T.; Morita, F.; Shimizu, K. Different responses to 5-fluoraouracil in mutagenicity and gene expression between two human lymphoblastoid cell lines with or without TP53 mutation. Acta Med. Okayama 2012, 66, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Godderis, L.; Thomas, R.; Hubbard, A.E.; Tabish, A.M.; Hoet, P.; Zhang, L.; Smith, M.T.; Veulemans, H.; McHale, C.M. Effect of chemical mutagens and carcinogens on gene expression profiles in human TK6 cells. PLoS ONE 2012, 7, e39205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehner, S.; Holzmann, K.; Speit, G. Characterization of formaldehyde’s genotoxic mode of action by gene expression analysis in TK6 cells. Arch. Toxicol. 2013, 87, 1999–2012. [Google Scholar] [CrossRef]
- Luan, Y.; Suzuki, T.; Palanisamy, R.; Takashima, Y.; Sakamoto, H.; Sakuraba, M.; Koizumi, T.; Saito, M.; Matsufuji, H.; Yamagata, K.; et al. Potassium bromate treatment predominantly causes large deletions, but not GC>TA transversion in human cells. Mutat. Res. 2007, 619, 113–123. [Google Scholar] [CrossRef]
- Piberger, A.L.; Krüger, C.T.; Strauch, B.M.; Schneider, B.; Hartwig, A. BPDE-induced genotoxicity: Relationship between DNA adducts, mutagenicity in the in vitro PIG-A assay, and the transcriptional response to DNA damage in TK6 cells. Arch. Toxicol. 2018, 92, 541–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buick, J.K.; Moffat, I.; Williams, A.; Swartz, C.D.; Recio, L.; Hyduke, D.R.; Li, H.-H.; Fornace, A.J.J.; Aubrecht, J.; Yauk, C.L. Integration of metabolic activation with a predictive toxicogenomics signature to classify genotoxic versus nongenotoxic chemicals in human TK6 cells. Environ. Mol. 2015, 56, 520–534. [Google Scholar] [CrossRef]
- Le Fevre, A.-C.; Boitier, E.; Marchandeau, J.-P.; Sarasin, A.; Thybaud, V. Characterization of DNA reactive and non-DNA reactive anticancer drugs by gene expression profiling. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2007, 619, 16–29. [Google Scholar] [CrossRef]
- Li, H.-H.; Hyduke, D.R.; Chen, R.; Heard, P.; Yauk, C.L.; Aubrecht, J.; Fornace, A.J., Jr. Development of a toxicogenomics signature for genotoxicity using a dose-optimization and informatics strategy in human cells. Environ. Mol. Mutagen. 2015, 56, 505–519. [Google Scholar] [CrossRef] [Green Version]
- Sakai, R.; Morikawa, Y.; Kondo, C.; Oka, H.; Miyajima, H.; Kubo, K.; Uehara, T. Combinatorial measurement of CDKN1A/p21 and KIF20A expression for discrimination of DNA damage-induced clastogenicity. Int. J. Mol. Sci. 2014, 15, 17256–17269. [Google Scholar] [CrossRef] [Green Version]
- Wilde, E.C.; Chapman, K.E.; Stannard, L.M.; Seager, A.L.; Brüsehafer, K.; Shah, U.-K.; Tonkin, J.A.; Brown, M.R.; Verma, J.R.; Doherty, A.T.; et al. A novel, integrated in vitro carcinogenicity test to identify genotoxic and non-genotoxic carcinogens using human lymphoblastoid cells. Arch. Toxicol. 2018, 92, 935–951. [Google Scholar] [CrossRef] [Green Version]
- OECD. Overview on Genetic Toxicology TGs; OECD Series on Testing and Assessment; OECD: Paris, France, 2017. [Google Scholar] [CrossRef]
- Phillips, D.H.; Arlt, V.M. Genotoxicity: Damage to DNA and its consequences. Mol. Clin. Environ. Toxicol. 2009, 99, 87–110. [Google Scholar] [CrossRef] [Green Version]
- Chrz, J.; Hošíková, B.; Svobodová, L.; Očadlíková, D.; Kolářová, H.; Dvořáková, M.; Kejlová, K.; Malina, L.; Jírová, G.; Vlková, A.; et al. Comparison of methods used for evaluation of mutagenicity/genotoxicity of model chemicals-parabens. Physiol. Res. 2020, 69, S661–S679. [Google Scholar] [CrossRef]
- Madia, F.; Kirkland, D.; Morita, T.; White, P.; Asturiol, D.; Corvi, R. EURL ECVAM Genotoxicity and Carcinogenicity Database of Substances Eliciting Negative Results in the Ames Test: Construction of the Database. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2020, 854–855, 503199. [Google Scholar] [CrossRef]
- Pistollato, F.; Madia, F.; Corvi, R.; Munn, S.; Grignard, E.; Paini, A.; Worth, A.; Bal-Price, A.; Prieto, P.; Casati, S.; et al. Current EU regulatory requirements for the assessment of chemicals and cosmetic products: Challenges and opportunities for introducing new approach methodologies. Arch. Toxicol. 2021, 95, 1867–1897. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Seo, J.-E.; Li, X.; Mei, N. Genetic toxicity assessment using liver cell models: Past, present, and future. J. Toxicol. Environ. Health Part B 2020, 23, 27–50. [Google Scholar] [CrossRef]
- Levy, D.D.; Zeiger, E.; Escobar, P.A.; Hakura, A.; van der Leede, B.-j.M.; Kato, M.; Moore, M.M.; Sugiyama, K.-i. Recommended criteria for the evaluation of bacterial mutagenicity data (Ames test). Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 848, 403074. [Google Scholar] [CrossRef] [PubMed]
- Punt, A.; Bouwmeester, H.; Blaauboer, B.J.; Coecke, S.; Hakkert, B.; Hendriks, D.F.G.; Jennings, P.; Kramer, N.I.; Neuhoff, S.; Masereeuw, R.; et al. New approach methodologies (NAMs) for human-relevant biokinetics predictions. Meeting the paradigm shift in toxicology towards an animal-free chemical risk assessment. ALTEX 2020, 37, 607–622. [Google Scholar] [CrossRef]
- Schaap, M.M.; Wackers, P.F.K.; Zwart, E.P.; Huijskens, I.; Jonker, M.J.; Hendriks, G.; Breit, T.M.; van Steeg, H.; van de Water, B.; Luijten, M. A novel toxicogenomics-based approach to categorize (non-)genotoxic carcinogens. Arch. Toxicol. 2015, 89, 2413–2427. [Google Scholar] [CrossRef]
- Wichard, J.D. In silico prediction of genotoxicity. Food Chem. Toxicol. 2017, 106, 595–599. [Google Scholar] [CrossRef]
- Ellinger-Ziegelbauer, H.; Stuart, B.; Wahle, B.; Bomann, W.; Ahr, H.J. Comparison of the expression profiles induced by genotoxic and nongenotoxic carcinogens in rat liver. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2005, 575, 61–84. [Google Scholar] [CrossRef]
- Hochstenbach, K.; van Leeuwen, D.M.; Gottschalk, R.W.; Gmuender, H.; Stolevik, S.B.; Nygaard, U.C.; Lovik, M.; Granum, B.; Namork, E.; van Loveren, H.; et al. Transcriptomic fingerprints in human peripheral blood mononuclear cells indicative of genotoxic and non-genotoxic carcinogenic exposure. Mutat. Res. 2012, 746, 124–134. [Google Scholar] [CrossRef] [PubMed]
- van Delft, J.H.; van Agen, E.; van Breda, S.G.; Herwijnen, M.H.; Staal, Y.C.; Kleinjans, J.C. Discrimination of genotoxic from non-genotoxic carcinogens by gene expression profiling. Carcinogenesis 2004, 25, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liska, D.J. The detoxification enzyme systems. Altern. Med. Rev. 1998, 3, 187–198. [Google Scholar] [PubMed]
- Song, M.-K.; Choi, H.-S.; Park, Y.-K.; Ryu, J.-C. Discovery of characteristic molecular signatures for the simultaneous prediction and detection of environmental pollutants. Environ. Sci. Pollut. Res. 2014, 21, 3104–3115. [Google Scholar] [CrossRef]
- Shah, U.-K.; Seager, A.L.; Fowler, P.; Doak, S.H.; Johnson, G.E.; Scott, S.J.; Scott, A.D.; Jenkins, G.J.S. A comparison of the genotoxicity of benzo[a]pyrene in four cell lines with differing metabolic capacity. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 808, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Ooka, M.; Lynch, C.; Xia, M. Application of in vitro metabolism activation in high-throughput screening. Int. J. Mol. Sci. 2020, 21, 8182. [Google Scholar] [CrossRef]
- Cox, J.A.; Fellows, M.D.; Hashizume, T.; White, P.A. The utility of metabolic activation mixtures containing human hepatic post-mitochondrial supernatant (S9) for in vitro genetic toxicity assessment. Mutagenesis 2015, 31, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Ebmeyer, J.; Braeuning, A.; Glatt, H.; These, A.; Hessel-Pras, S.; Lampen, A. Human CYP3A4-mediated toxification of the pyrrolizidine alkaloid lasiocarpine. Food Chem. Toxicol. 2019, 130, 79–88. [Google Scholar] [CrossRef]
- Brimer, P.A.; Tan, E.-L.; Hsie, A.W. Effect of metabolic activation on the cytotoxicity and mutagenicity of 1,2-dibromoethane in the CHO/HGPRT system. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1982, 95, 377–388. [Google Scholar] [CrossRef]
- Ku, W.W.; Bigger, A.; Brambilla, G.; Glatt, H.; Gocke, E.; Guzzie, P.J.; Hakura, A.; Honma, M.; Martus, H.-J.; Obach, R.S.; et al. Strategy for genotoxicity testing—Metabolic considerations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2007, 627, 59–77. [Google Scholar] [CrossRef]
- Yauk, C.L.; Buick, J.K.; Williams, A.; Swartz, C.D.; Recio, L.; Li, H.-H.; Fornace, A.J., Jr.; Thomson, E.M.; Aubrecht, J. Application of the TGx-28.65 transcriptomic biomarker to classify genotoxic and non-genotoxic chemicals in human TK6 cells in the presence of rat liver S9. Environ. Mol. Mutagen. 2016, 57, 243–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillouzo, A.; Corlu, A.; Aninat, C.; Glaise, D.; Morel, F.; Guguen-Guillouzo, C. The human hepatoma HepaRG cells: A highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem. Biol. Interact. 2007, 168, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 mutation status in human cancer cell lines: A reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Millau, J.-F.; Mai, S.; Bastien, N.; Drouin, R. p53 functions and cell lines: Have we learned the lessons from the past? BioEssays 2010, 32, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.N.; Li, Y.; Nakamoto, K.; Subileau, E.-A.; Steen, D.; Zhong, X.-B. A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennen, D.G.J.; Magkoufopoulou, C.; Ketelslegers, H.B.; van Herwijnen, M.H.M.; Kleinjans, J.C.S.; van Delft, J.H.M. Comparison of HepG2 and HepaRG by whole-genome gene expression analysis for the purpose of chemical hazard identification. Toxicol. Sci. 2010, 115, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Berger, B.; Donzelli, M.; Maseneni, S.; Boess, F.; Roth, A.; Krähenbühl, S.; Haschke, M. Comparison of liver cell models using the basel phenotyping cocktail. Front. Pharmacol. 2016, 7, 443. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-H.; Yauk, C.L.; Chen, R.; Hyduke, D.R.; Williams, A.; Frötschl, R.; Ellinger-Ziegelbauer, H.; Pettit, S.; Aubrecht, J.; Fornace, A.J. TGx-DDI, a transcriptomic biomarker for genotoxicity hazard assessment of pharmaceuticals and environmental chemicals. Front. Big Data 2019, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Buick, J.K.; Williams, A.; Gagné, R.; Swartz, C.D.; Recio, L.; Ferguson, S.S.; Yauk, C.L. Flow cytometric micronucleus assay and TGx-DDI transcriptomic biomarker analysis of ten genotoxic and non-genotoxic chemicals in human HepaRG™ cells. Genes Environ. 2020, 42, 5. [Google Scholar] [CrossRef]
- Sasaki, J.C.; Allemang, A.; Bryce, S.M.; Custer, L.; Dearfield, K.L.; Dietz, Y.; Elhajouji, A.; Escobar, P.A.; Fornace, A.J., Jr.; Froetschl, R.; et al. Application of the adverse outcome pathway framework to genotoxic modes of action. Environ. Mol. Mutagen. 2020, 61, 114–134. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.L.; Levine, A.J. The p53 pathway: Positive and negative feedback loops. Oncogene 2005, 24, 2899–2908. [Google Scholar] [CrossRef] [Green Version]
- Josse, R.; Dumont, J.; Fautrel, A.; Robin, M.-A.; Guillouzo, A. Identification of early target genes of aflatoxin B1 in human hepatocytes, inter-individual variability and comparison with other genotoxic compounds. Toxicol. Appl. Pharmacol. 2012, 258, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Lereau, M.; Gouas, D.; Villar, S.; Besaratinia, A.; Hautefeuille, A.; Berthillon, P.; Martel-Planche, G.; Nogueira da Costa, A.; Ortiz-Cuaran, S.; Hantz, O.; et al. Interactions between hepatitis B virus and aflatoxin B(1): Effects on p53 induction in HepaRG cells. J. Gen. Virol. 2012, 93, 640–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smart, D.J.; Helbling, F.R.; Verardo, M.; Huber, A.; McHugh, D.; Vanscheeuwijck, P. Development of an integrated assay in human TK6 cells to permit comprehensive genotoxicity analysis in vitro. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2020, 849, 503129. [Google Scholar] [CrossRef] [PubMed]
- Amundson, S.A.; Do, K.T.; Vinikoor, L.; Koch-Paiz, C.A.; Bittner, M.L.; Trent, J.M.; Meltzer, P.; Fornace, A.J., Jr. Stress-specific signatures: Expression profiling of p53 wild-type and -null human cells. Oncogene 2005, 24, 4572–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doktorova, T.Y.; Yildirimman, R.; Ceelen, L.; Vilardell, M.; Vanhaecke, T.; Vinken, M.; Ates, G.; Heymans, A.; Gmuender, H.; Bort, R.; et al. Testing chemical carcinogenicity by using a transcriptomics HepaRG-based model? EXCLI J. 2014, 13, 623–637. [Google Scholar] [PubMed]
- Boehme, K.; Dietz, Y.; Hewitt, P.; Mueller, S.O. Genomic profiling uncovers a molecular pattern for toxicological characterization of mutagens and promutagens in vitro. Toxicol. Sci. 2011, 122, 185–197. [Google Scholar] [CrossRef]
- Jetten, M.J.A.; Kleinjans, J.C.S.; Claessen, S.M.; Chesné, C.; van Delft, J.H.M. Baseline and genotoxic compound induced gene expression profiles in HepG2 and HepaRG compared to primary human hepatocytes. Toxicol. Vitr. 2013, 27, 2031–2040. [Google Scholar] [CrossRef]
- Labib, S.; Williams, A.; Guo, C.H.; Leingartner, K.; Arlt, V.M.; Schmeiser, H.H.; Yauk, C.L.; White, P.A.; Halappanavar, S. Comparative transcriptomic analyses to scrutinize the assumption that genotoxic PAHs exert effects via a common mode of action. Arch. Toxicol. 2016, 90, 2461–2480. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Yum, Y.N.; Kim, S.C.; Kim, Y.; Lim, J.; Lee, W.J.; Koo, K.H.; Kim, J.H.; Kim, J.E.; Lee, W.S.; et al. Distinguishing between genotoxic and non-genotoxic hepatocarcinogens by gene expression profiling and bioinformatic pathway analysis. Sci. Rep. 2013, 3, 2783. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Minowa, Y.; Morikawa, Y.; Kondo, C.; Maruyama, T.; Kato, I.; Nakatsu, N.; Igarashi, Y.; Ono, A.; Hayashi, H.; et al. Prediction model of potential hepatocarcinogenicity of rat hepatocarcinogens using a large-scale toxicogenomics database. Toxicol. Appl. Pharmacol. 2011, 255, 297–306. [Google Scholar] [CrossRef]
- Cunha, I.W.; Carvalho, K.C.; Martins, W.K.; Marques, S.M.; Muto, N.H.; Falzoni, R.; Rocha, R.M.; Aguiar, S.; Simoes, A.C.Q.; Fahham, L.; et al. Identification of genes associated with local aggressiveness and metastatic behavior in soft tissue tumors. Transl. Oncol. 2010, 3, 23–32. [Google Scholar] [CrossRef]
- Bakshi, D.; Katoch, A.; Chakraborty, S.; Shah, R.; Sharma, B.; Bhat, A.; Verma, S.; Bhat, G.R.; Nagpal, A.; Vaishnavi, S.; et al. ANKLE1 as new hotspot mutation for breast cancer in indian population and has a role in DNA damage and repair in mammalian cells. Front. Genet. 2020, 11, 609758. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tian, T.; Li, Z.Y.; Wang, C.Y.; Deng, R.; Deng, W.Y.; Yang, A.K.; Chen, Y.F.; Li, H. FSCN1 is an effective marker of poor prognosis and a potential therapeutic target in human tongue squamous cell carcinoma. Cell Death Dis. 2019, 10, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Zhao, Z.; Duan, X.; Chen, P.; Peng, Z.; Qiu, H. FSCN1 predicts survival and is regulated by a PI3K-dependent mechanism in renal cell carcinoma. J. Cell Physiol. 2018, 233, 4748–4758. [Google Scholar] [CrossRef] [PubMed]
- di Pietro, M.; Marra, G.; Cejka, P.; Stojic, L.; Menigatti, M.; Cattaruzza, M.S.; Jiricny, J. Mismatch repair-dependent transcriptome changes in human cells treated with the methylating agent N-methyl-N’-nitro-N-nitrosoguanidine. Cancer Res. 2003, 63, 8158–8166. [Google Scholar]
- Luo, W.; Fan, W.; Xie, H.; Jing, L.; Ricicki, E.; Vouros, P.; Zhao, L.P.; Zarbl, H. Phenotypic anchoring of global gene expression profiles induced by N-hydroxy-4-acetylaminobiphenyl and benzo[a]pyrene diol epoxide reveals correlations between expression profiles and mechanism of toxicity. Chem. Res. Toxicol. 2005, 18, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC. A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Stacklies, W.; Redestig, H.; Scholz, M.; Walther, D.; Selbig, J. pcaMethods—a bioconductor package providing PCA methods for incomplete data. Bioinformatics 2007, 23, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kreuzer, K.; Sprenger, H.; Braeuning, A. Comparative Analysis of Transcriptional Responses to Genotoxic and Non-Genotoxic Agents in the Blood Cell Model TK6 and the Liver Model HepaRG. Int. J. Mol. Sci. 2022, 23, 3420. https://doi.org/10.3390/ijms23073420
Kreuzer K, Sprenger H, Braeuning A. Comparative Analysis of Transcriptional Responses to Genotoxic and Non-Genotoxic Agents in the Blood Cell Model TK6 and the Liver Model HepaRG. International Journal of Molecular Sciences. 2022; 23(7):3420. https://doi.org/10.3390/ijms23073420
Chicago/Turabian StyleKreuzer, Katrin, Heike Sprenger, and Albert Braeuning. 2022. "Comparative Analysis of Transcriptional Responses to Genotoxic and Non-Genotoxic Agents in the Blood Cell Model TK6 and the Liver Model HepaRG" International Journal of Molecular Sciences 23, no. 7: 3420. https://doi.org/10.3390/ijms23073420
APA StyleKreuzer, K., Sprenger, H., & Braeuning, A. (2022). Comparative Analysis of Transcriptional Responses to Genotoxic and Non-Genotoxic Agents in the Blood Cell Model TK6 and the Liver Model HepaRG. International Journal of Molecular Sciences, 23(7), 3420. https://doi.org/10.3390/ijms23073420